Nature:基于三代测序+光学图谱+Hi-C构建高质量藜麦基因组
【字体: 大 中 小 】 时间:2017年02月21日 来源:基因有限公司
编辑推荐:
尽管藜麦具有农业潜力,但仍然未被充分利用。人们继续努力改良其重要的农业性状以拓展藜麦在全球范围生产。为了加速其改良,本文作者报道了异源四倍体的藜麦基因组,并找到了可能调控种子中皂苷含量的基因。

图一 野生藜麦
研究背景:提到藜麦Chenopodium quinoa (quinoa),很多人都比较陌生,然而早在7000多年前南美的安第斯高山地区,藜麦就已经被人类种植并在数个世纪后成为印加帝国众所周知的“母亲的谷物”。藜麦已经适应了安第斯高原环境(海拔高于3500米),并进化出了对多种非生物逆境的适应能力,被认为是重要的可改善世界粮食安全的作物。藜麦因为其种子的营养价值已经引起全球的关注,它不含谷蛋白,升糖指数更低,并含有非常均衡的氨基酸,纤维,脂肪,碳水化合物,维生素和矿物质等营养物质。并且可以在不适于主要粮食作物生长的土地上种植。尽管藜麦具有农业潜力,但仍然未被充分利用。人们继续努力改良其重要的农业性状以拓展藜麦在全球范围生产。为了加速其改良,本文作者报道了异源四倍体的藜麦基因组,并找到了可能调控种子中皂苷含量的基因。

研究策略与结果比较:
本文作者报道了他们采用PacBio三代测序、Bionano光学图谱、Hi-C技术结合遗传图谱组装的高质量染色体级别的藜麦参考基因组序列并发表在最新的Nature杂志上。
事实上,在2016年,也有日本的研究团队对藜麦基因组进行过测序,采用的是短读长的二代测序技术结合低深度的PacBio测序数据,因此最后只得到了草图(draft),并非完整的参考基因组,文章仅发在了DNA Research(IF:5.267)杂志上。时隔半年,为何此次对藜麦的测序可以发到Nature(IF:38.138)的article?下面我们来具体了解一下两篇文章有何区别?
藜麦的基因组(Chenopodium quinoa, 2n=4x=36)预计在1.45-1.50Gb左右,本文中采用PacBio三代测序+Bionano光学图谱+Hi-C的经典策略,总共组装得到了1.39Gb的基因组序列,总共包含3,486个scaffold,Scaffold N50大小为3.84Mb,90%的基因组包含于439个scaffold中。而之前发表于DNA Research,基于短读长得到的草图获得了24,000多个scaffold,丢失了25%以上的序列。
具体比较如下:
|
发表期刊 |
Nature (Feb,2017) |
DNA Research(Jul, 2016) |
|
影响因子 |
38.138 |
5.267 |
|
组装策略 |
PacBio三代测序数据+Bionano光学图谱+Hi-C结合遗传图谱组装 |
Illumina Hiseq 2500数据为主,PacBio数据用于gap-closing和下一步的scaffolding |
|
数据量 |
PacBio 20kb文库,P6-C4试剂,100个SMRTCell 总共75Gb数据,平均读长12,444bp |
Illumina数据290.8 Gb,深度196x PacBio数据45.8Gb,深度31x |
|
Contig N50 |
1.66 Mb |
14,505 bp |
|
Scaffold数量 |
3,486 |
24,845 |
|
Scaffold N50 |
3.84 Mb |
86,941 bp |
|
最大scaffold |
23.8 Mb |
0.64 Mb |
|
组装碱基数 |
1.39 Gb(4.56% missing) |
1.087Gb(>25% missing) |
从比较数据不难看出,对于藜麦这样的复杂异源四倍体基因组,64%的序列是重复序列,包含大量的长末端重复(LTR)转座因子。基于PacBio+Bionano+Hi-C的组装策略远优于短读长测序技术为主的组装策略,无论在基因组覆盖度,还是Contig N50和Scaffold N50等组装参数上都有大幅提升。在已有二代测序已发表基因组草图的情况下,仍然能够发表于Nature杂志上。
藜麦的进化历史:
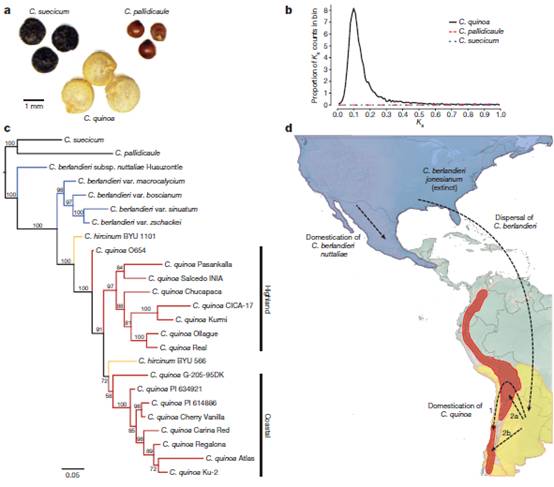
图二 藜麦进化史
藜麦是由祖源的A和B两个二倍体品种杂交而来。之前的单基因测序研究鉴定到种质库中,北美和欧亚两个二倍体分别为A亚基因组和B亚基因组的候选来源,后来在北美某处发生了杂交。为了进一步了解藜麦的基因组结构和进化,作者对A基因组二倍体C. pallidicaule和B基因组二倍体C. suecicum进行了测序、组装和注释。藜麦中同源基因对的很大比例在每个同义位点表现出相似的同义替代率,表明全基因组的复制事件。作者估计重组大约发生在3.3-6.3百万年前。进化树分析表明北美C. berlandieri是物种综合的基本成员。藜麦被认为是在一次单独事件中从C. hircinum驯化而来。而作者的测序数据表明藜麦可能分别在高原和沿海环境被独立驯化。作者从这些登记样本和参考藜麦基因组中找到了总共7,809,381个SNP,包括2,668,694个藜麦特异的SNP。这将有助于评估其遗传多样性,以及鉴定有有价值的性状相关的基因组区域。
亚基因组特点分析:
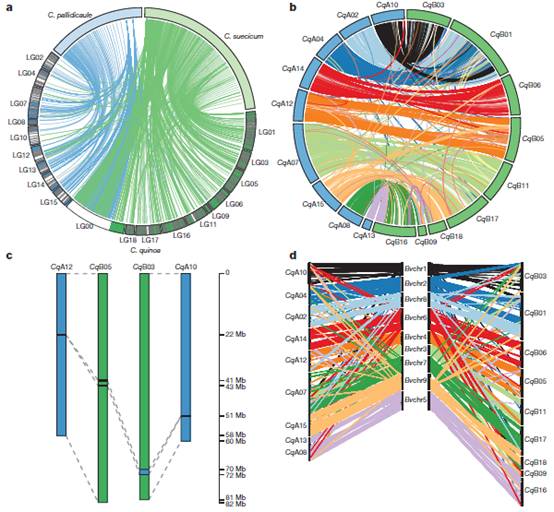
图三:亚基因组的鉴定与特点
通过把来自C. pallidicaule 和C. suecicum 的测序结果mapping到藜麦的scaffold组装结果上,进行BLASTN把每个二倍体与藜麦的scaffold比对,发现分别有156个和410个藜麦scaffold比对到A基因组和B基因组上(总共202.6Mb和646.3Mb)。5,807个同源基因对在染色体上的定位揭示其与A,B亚基因组的高度共线性。
皂苷合成的潜在机制:
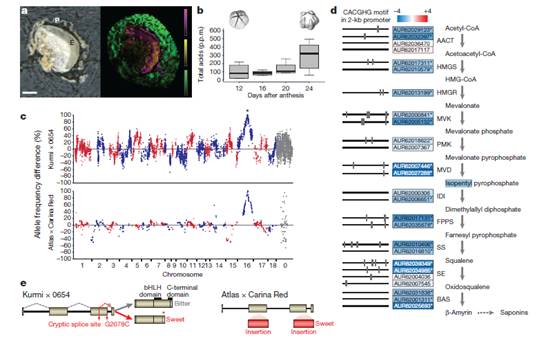
图四:皂苷合成的候选基因
a. 藜麦种子的外皮中的皂苷质谱分析成像
b. 在种子发育期间通过总酸量测定检测皂苷的积累
c. 群体中,甜味后代与苦味后代相比的等位基因频率的比例差异
d. 皂苷生物合成途径,注明了途径中催化每一步反应的酶和编码每个酶的基因ID
e. 苦味和甜味品种中的TSARL1基因模式
藜麦种子中含有皂苷(saponins)。尽管这对于植物生长有益(比如阻止食草性动物),但是为了人类能够食用,必须去除掉,因为其具有溶血性和苦味。但要去除皂苷,费用太高,耗水量大,也会降低种子的营养价值。作者发现皂苷集中于开花期后20-24天的果皮中,最终占到种子总质量的4%(w/w)。
天然存在的甜味藜麦含有很低的皂苷水平,尽管潜在的调控基因尚不清楚。为了找到这些基因,作者进行了两个杂交分离群体的连锁图谱和BSA的分析:Kurmi (甜味) × 0654 (苦味),和Atlas (甜味) × Carina Red (苦味)。与其他群体中得到的结果相同,这些群体中的分离比表明种子中皂苷的存在是由一个单独的基因控制的。皂苷的有无与种皮厚度差异相关。苦味的品种比甜味品种有更厚的种皮。
转录组分析:
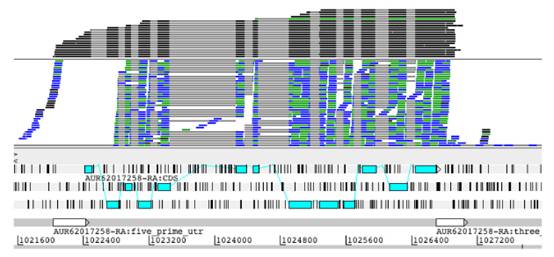
图五:用RNA-Seq和Iso-Seq分别分析转录组选择性剪切情况
图中上部显示的是PacBio Iso-Seq转录组测序结果,中间部分显示的是Illumina RNA-Seq测序结果。上两部分的灰色线条表示内含子区域。底部显示的是AUR62017258基因所在的染色体位置。明显可以看出,PacBio的长读长技术无需拼接用于全长转录本测序,可以在一个reads中完全覆盖从5′非翻译区,所有外显子和3′非翻译区。而短读长技术则需要进行组装。
小结:
藜麦作为一种新兴的作物,因为其营养丰富,耐受多种非生物胁迫,被认为对改善全球粮食安全有重大潜力。高质量的基因组组装结果将加速其遗传改良。PacBio三代测序结合Bionano光学图谱技术和Hi-C技术可以完美组装复杂基因组。同时,PacBio长读长reads可以轻松获得完整的转录本全长无需拼接。本文再次证实其在基因组组装,转录本选择性剪切等应用中的优势。
欢迎索取PacBio三代测序和Bionano光学图谱的更多资料
原文信息:
Jarvis, David E. "The Genome of Chenopodium quinoa." Nature (2016).
文献全文请参考:
http://www.nature.com/nature/journal/vaop/ncurrent/full/nature21370.html
基因有限公司作为Pacific Biosciences公司中国区独家代理商,以及BioNano公司中国区代理商。自2011年以来将PacBio第三代单分子实时测序技术引入国内,一直为国内用户提供专业的三代测序系统、单分子光学图谱系统的安装培训,技术支持,应用培训与售后维护工作,赢得客户的一致好评与信任。基因有限公司将一如既往的支持越来越多的PacBio及BioNano用户。




